Regolamento sui Dispositivi Medici
Regolamento (UE) 2017/745

Requisiti MDR (UE)
Fresenius Medical Care è impegnata a garantire che i propri dispositivi medici siano conformi ai requisiti MDR applicabili secondo le rispettive tempistiche di transizione.
Sono molte le domande che sorgono in merito all’MDR, in particolare per quanto riguarda:
-
Il suo impatto sulla disponibilità continuativa dei prodotti di Fresenius Medical Care
-
I dettagli sulla sua applicabilità per i clienti e gli utilizzatori dei nostri dispositivi medici
Data la vastità e la complessità del nuovo MDR, i nuovi requisiti vengono attuati in base a programmi di implementazione dedicati e controllati dal Management in stretta collaborazione con il nostro Ente notificato (un elenco di tutti gli Enti certificati designati secondo l’MDR si trova nella sezione dei link più in basso*).
Le domande e le risposte che seguono forniscono informazioni rilevanti in merito all’implementazione dell’MDR da parte di Fresenius Medical Care e sulle sue conseguenze per i clienti e gli utilizzatori dei nostri dispositivi medici.
Domande e risposte
Fresenius Medical Care soddisfa i requisiti MDR?
La conformità del Sistema di gestione della qualità di Fresenius Medical Care ai requisiti MDR applicabili è stata valutata e confermata nel corso di audit esterni condotti dall’Ente certificato nel 2019. Da allora, siamo soggetti ad audit di sorveglianza periodici sulla conformità al regolamento MDR.
Un prerequisito non meno importante per il conseguimento della certificazione MDR è rappresentato dalla verifica dei prodotti in termini di conformità ai requisiti del regolamento MDR. Dopo una valutazione positiva da parte dell’Ente certificato, la classe I di prodotti è stata inclusa nell’ambito della certificazione MDR. Il numero di prodotti conformi all’MDR aumenterà costantemente nel corso dei prossimi anni, in linea con le disposizioni per la transizione all’MDR.
Il rispettivo certificato UE ai sensi dell’MDR, emesso dall’Ente certificato, verrà aggiornato di volta in volta con le categorie di prodotti corrispondenti.

Tempistiche per la transizione al Regolamento Dispositivi Medici (UE) 2017/745 tenuto conto del posticipo della data di applicazione dell’MDR così come disposto dal Regolamento (UE) 2020/561
Quali sono le tempistiche di transizione dalla precedente Direttiva Dispositivi Medici (MDD) al nuovo regolamento MDR applicabili a Fresenius Medical Care?
Per le tempistiche generali relative alle disposizioni sulla transizione all’MDR vedere la figura.
Per classi di rischio specifiche di dispositivi medici, potranno essere applicate tempistiche più stringenti, in accordo ai regolamenti applicabili.
Tuttavia, le tempistiche relative all’MDR sono soggette a revisione e a possibili modifiche se ritenute necessarie dalla Commissione UE. Principali esempi:
- A causa della crisi del COVID-19, la data di applicazione dell’MDR è stata posticipata di un anno dalla Commissione UE, al 26 maggio 2021 (Regolamento (UE) 2020/561).
- La tempistica di transizione per gli obblighi di certificazione di prodotti specifici di classe I è stata posticipata da maggio 2020 a maggio 2024. I gruppi di prodotti interessati sono, ad es., le applicazioni software per dispositivi medici in corso di riclassificazione (Corrigendum al Regolamento (UE) 2017/745, del 25 novembre 2019)
Fonte: Versione modificata dell’infografica della Commissione UE
Quand’è che tutti i prodotti Fresenius Medical Care interessati saranno conformi ai nuovi requisiti MDR?
I nostri dispositivi medici saranno immessi sul mercato UE conformemente ai requisiti UE applicabili nelle tempistiche definite dalla legge per la transizione dalla precedente direttiva MDD al regolamento MDR.
Fino a maggio 2024, i prodotti che sono stati certificati ai sensi della direttiva MDD potranno ancora, in determinate circostanze, essere commercializzati dopo la piena entrata in vigore dell’MDR, purché i loro certificati CE siano validi.
Pertanto, durante il periodo di transizione Fresenius Medical Care immetterà sul mercato sia dispositivi medici con certificazione MDD che dispositivi medici con certificazione MDR e, fino a maggio 2024, i prodotti Fresenius Medical Care con certificazione MDD verranno progressivamente certificati ai sensi del regolamento MDR. In questa fase potranno rendersi necessari degli adattamenti alla gamma di prodotti FME.
Fino a maggio 2025 potrà ancora proseguire la vendita di prodotti certificati MDD fino ad esaurimento delle scorte, che saranno riassortite con prodotti certificati MDR.
Quali sono le modifiche previste per l’etichettatura dei prodotti certificati MDR?
I prodotti certificati MDR devono avere un identificativo univoco del dispositivo (UDI). L’UDI è costituito da una serie di caratteri numerici o alfanumerici che consente di identificare un prodotto specifico sul mercato.
Per i prodotti certificati MDR sono necessari dei simboli specifici; vedere la prima figura per alcuni esempi.
La codifica UDI sui dispositivi conformi al regolamento MDR non sostituisce il codice articolo, ma viene utilizzata per l’identificazione e la tracciabilità del prodotto lungo l’intero ciclo di vita del dispositivo*.
La seconda figura mostra un esempio di etichetta generica conforme al regolamento MDR.
Esempio di simboli MDR
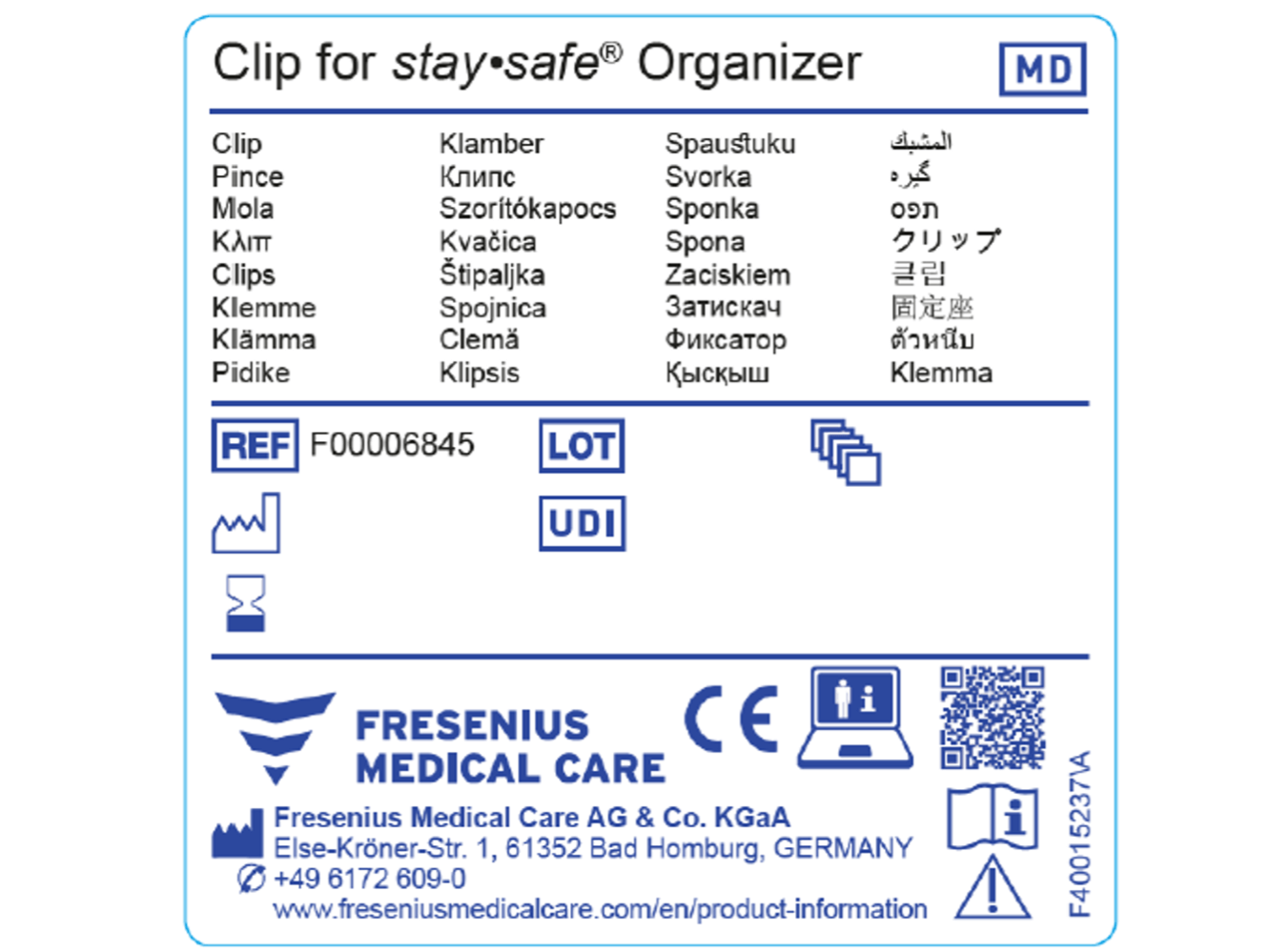
Esempio di etichetta MDR
Che livello di tracciabilità del prodotto deve essere garantito dai nostri clienti in merito ai nostri dispositivi medici dopo l’entrata in vigore del requisito relativo all’etichettatura UDI?
I prodotti certificati MDR devono avere un identificativo univoco del dispositivo (UDI). L’UDI è costituito da una serie di caratteri numerici o alfanumerici che consente di identificare un prodotto specifico sul mercato.
Gli obblighi rilevanti per i clienti sono descritti nelle schede informative della Commissione UE (vedere il link nella sezione seguente) per:
- Identificativo univoco del dispositivo (UDI)
- Rappresentanti autorizzati, distributori e importatori
- Professionisti sanitari e strutture sanitarie
Qual è la rilevanza della banca dati EUDAMED?
La banca dati europea sui dispositivi medici (EUDAMED) è un database gestito dalla Commissione UE per l’archiviazione delle informazioni riguardanti i dispositivi medici.
La finalità dell’EUDAMED è di rafforzare la sorveglianza del mercato fornendo alle autorità competenti un rapido accesso alle informazioni relative a fabbricanti, rappresentanti autorizzati, dispositivi medici, certificati e dati sulla vigilanza, condividere le informazioni sui dati delle indagini cliniche, nonché contribuire all’applicazione uniforme dei requisiti normativi, in particolare per quanto riguarda i requisiti in materia di registrazione.
Tutti i fabbricanti e tutti i dispositivi medici da immettere sul mercato europeo saranno registrati in EUDAMED. I fabbricanti (come Fresenius Medical Care) devono assicurarsi che tutti i dati forniti alla banca dati EUDAMED siano corretti e aggiornati. I dati comprendono: dati relativi ai prodotti, dati provenienti dai diversi rapporti di vigilanza del mercato e dati riguardanti la registrazione degli Operatori Economici.
Fresenius Medical Care deve occuparsi dell’adempimento dei requisiti in materia di registrazione in EUDAMED e di rendicontazione entro le tempistiche definite dalla legge.
Istruzioni per l’uso scaricabili
Le istruzioni per l’uso (IFU) dei prodotti Fresenius Medical Care con certificazione MDR, saranno fornite online, oltre la versione stampata. Le istruzioni per l’uso dei prodotti con certificazione MDR sono reperibili qui.
Informazioni di background sull’MDR
Informazioni e schede informative fornite dalla Commissione UE
Per trovare i seguenti documenti eseguire una ricerca rapida per:
- Regolamento (UE) 2017/745 (MDR)
- Sito Web della Commissione UE con Informazioni generali sull’MDR.
- Elenco di tutti gli Enti notificati designati secondo l’MDR
Per altri link vedere:
- Panoramica su varie schede informative e guide (vedere anche più in basso)
- Schede informative per fabbricanti di dispositivi medici
- Scheda informativa sull’UDI (formato PDF)
- Scheda informativa per professionisti sanitari e strutture sanitarie
- Scheda informativa per autorità competenti in Paesi extra-UE/AEE
Informazioni e schede informative da altre fonti
Associazioni industriali e autorità pubbliche per informazioni generali sull’MDR:
*Schede informative fornite dalla Commissione UE in merito all’UDI, per Rappresentanti autorizzati / Distributori / Importatori e Professionisti Sanitari